Complete, closed bacterial genomes from microbiomes using nanopore sequencing
Abstract
Microbial genomes can be assembled from short-read sequencing data, but the assembly contiguity of these metagenome-assembled genomes is constrained by repeat elements. Correct assignment of genomic positions of repeats is crucial for understanding the effect of genome structure on genome function.
We applied nanopore sequencing and our workflow, named Lathe, which incorporates long-read assembly and short-read error correction, to assemble closed bacterial genomes from complex microbiomes.
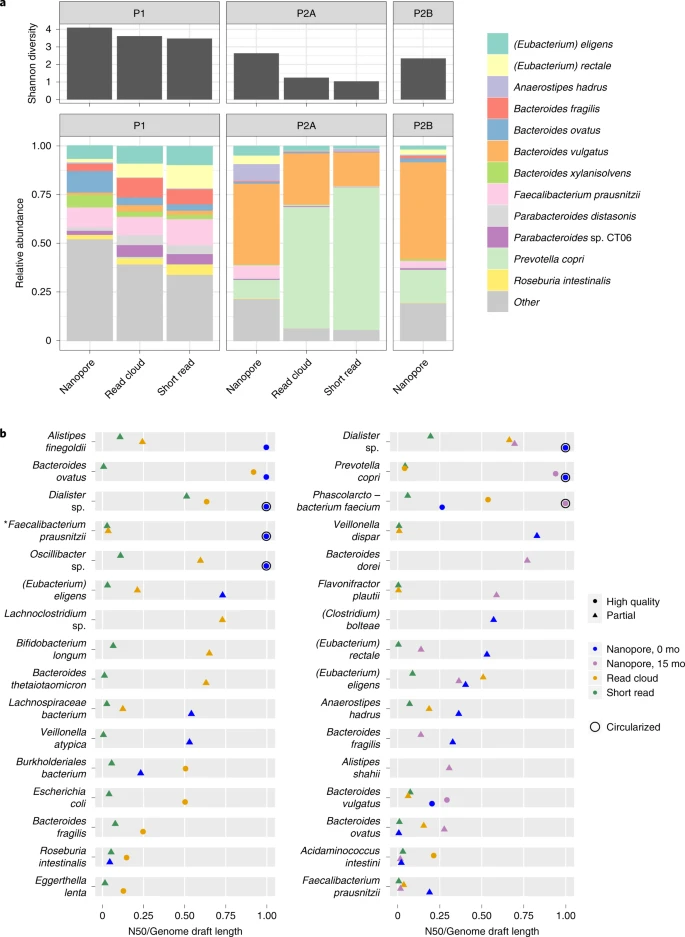
We validated our approach with a synthetic mixture of 12 bacterial species. Seven genomes were completely assembled into single contigs and three genomes were assembled into four or fewer contigs. Next, we used our methods to analyze metagenomics data from 13 human stool samples. We assembled 20 circular genomes, including genomes of Prevotella copri and a candidate Cibiobacter sp. Despite the decreased nucleotide accuracy compared with alternative sequencing and assembly approaches, our methods improved assembly contiguity, allowing for investigation of the role of repeat elements in microbial function and adaptation.
Content
果然他們也是卡在萃取高品質的DNA,是說HMW為何需要簡稱XD
However, the application of long-read methods to analyze gut microbiomes has been hindered by the lack of efficient methods to extract high molecular weight (HMW) DNA from stool.
然後就提出一套優質的萃取步驟還有生資分析流程
enzymatic degradation of the cell wall with a cocktail of lytic enzymes, then phenol-chloroform extraction, followed by RNAse A and Proteinase K digestion, gravity column purification and solid phase reversible immobilization (SPRI) size selection.
Supplementary Figure 1 Overview of the molecular and informatic workflow steps.
Bioinformatic workflow: Lathe (原來可以取綽號?)
看不太到有啥新穎的演算法,都是使用一些既有的工具而已
應該是特別在調整參數吧
比較精華的應該就是有趣處理misassmbly的reads吧
To detect misassemblies, Lathe searches for locations in the assembly spanned across by one or zero long reads, indicating either a total lack of support for true contiguity or support from only a single possibly chimeric read. It does this by breaking the genome into windows smaller than the average read length, then measuring coverage within each window from reads spanning the entire window. With no misassembly, an assembly produced from a given readset will have all windows spanned by the assembled reads. A misassembly within a given window will cause read alignments to be soft-clipped at the misassembly breakpoint, preventing read alignments from spanning across the breakpoint and therefore the window. Contigs are then broken at identified misassembled sites before final output generation.
也很佛心的有把workflow上傳github,還是使用snakemake把工具整合在一起
雖然我也不太了解snakemake
Lathe can be found at https://github.com/bhattlab/lathe/.
再來就菌種鑑定跟註解,也是滿通用的工具
Genomes were compared to reference sequences by alignment with Mummer.
Long and short reads were taxonomically classified with Kraken
Supplementary Figure 9 Taxonomic composition across extraction and sequencing methods.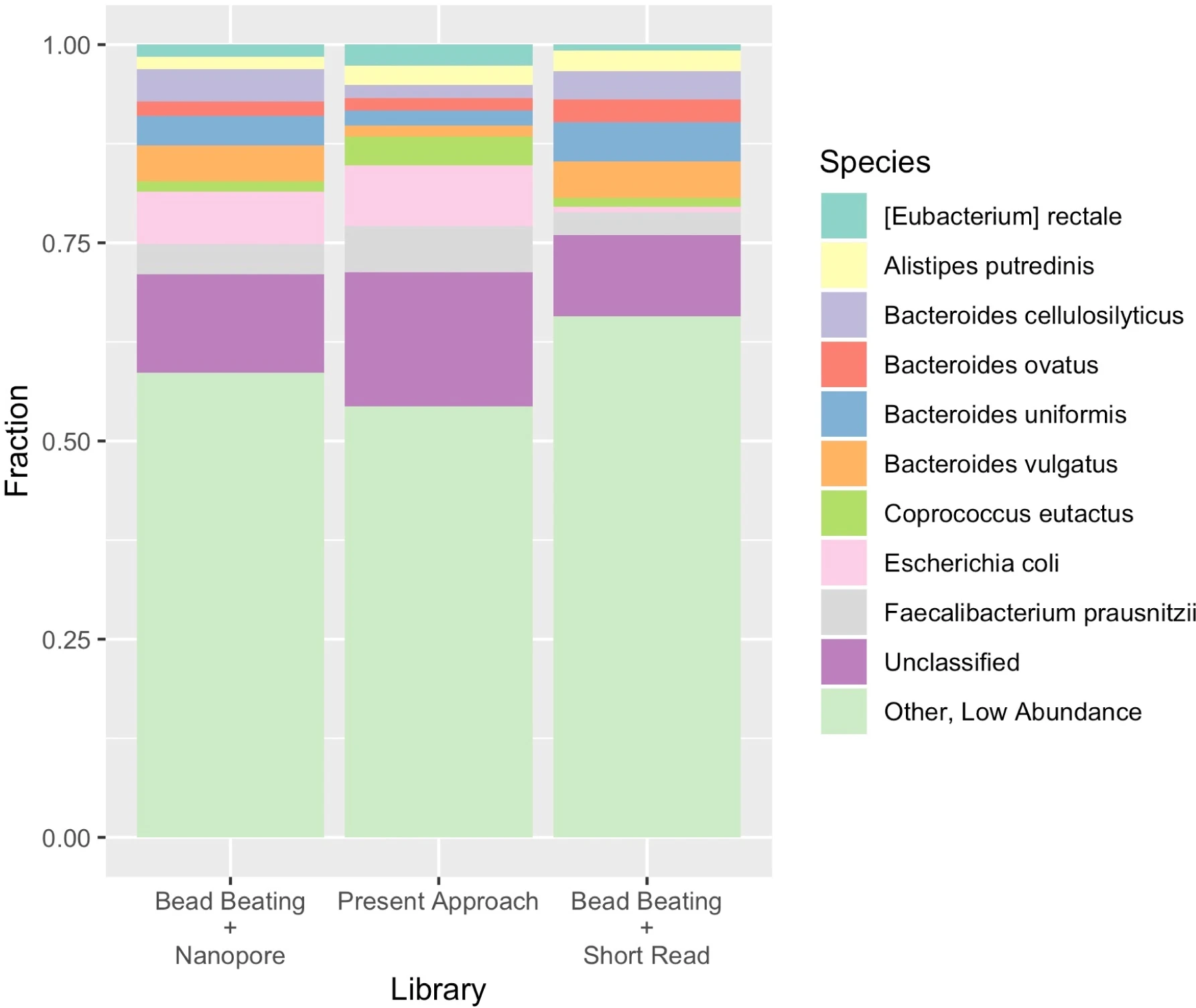
這R可以做耶
Shannon diversity was calculated with vegan.
果然也有提到reads校正的問題
We note that classifiers developed for short reads of uniform length do not correct for the variable read length of long reads, counting relative read counts and not relative number of bases sequenced, which may slightly bias relative abundance results.
再來就基本的註解流程了
rRNA presence was determined with Barrnap v.0.9 (ref. 37). Gene count and insertion sequence transposase count was determined with Prokka v.1.13.3 (ref. 16). For P. copri, additional insertion sequence locations were determined by mapping previously identified P. copri insertion sequences5 to the circular genome. Putative phage regions were identified with PHASTER38. Figures were generated with ggplot2 v.3.2.1 (ref. 39).
他一樣用snakemake整合,或許workflow的腳本也都可以用這工具整合耶,目前都用bash寫腳本而已
再研究看看好處好了
Downstream analysis workflows can be found at https://github.com/bhattlab/metagenomics_workflows/.
Novel species identification這塊看起來也就blast database啊
到底是怎麼決定新物種
Classifications for the unknown genome assembled in sample P1 and shown in Fig. 2 and Table 1 as F. prausnitzii were attempted with BLAST v.2.9.0 (ref. 40) against the NCBI Genbank database, 16S identification with Barrnap v.0.9 (ref. 37) and BLAST against the Ribosomal Database Project database and NCBI 16S Archaeal and Bacterial database, and Kraken2 classification35. Genome sequences were compared to the assembled genome draft by alignment and post-processing with mummer.
Fig. 2: Per-organism assembly contiguity, diversity and taxonomic read composition in two healthy human stool microbiomes.